Data Storage¶
This page describes the design of the database that is/will be used in order to store all necessary pieces information that are obtained from the "stand-alone" ProCKSI/Comparison and ProCKSI/Consensus applications (see DataStandardisation).
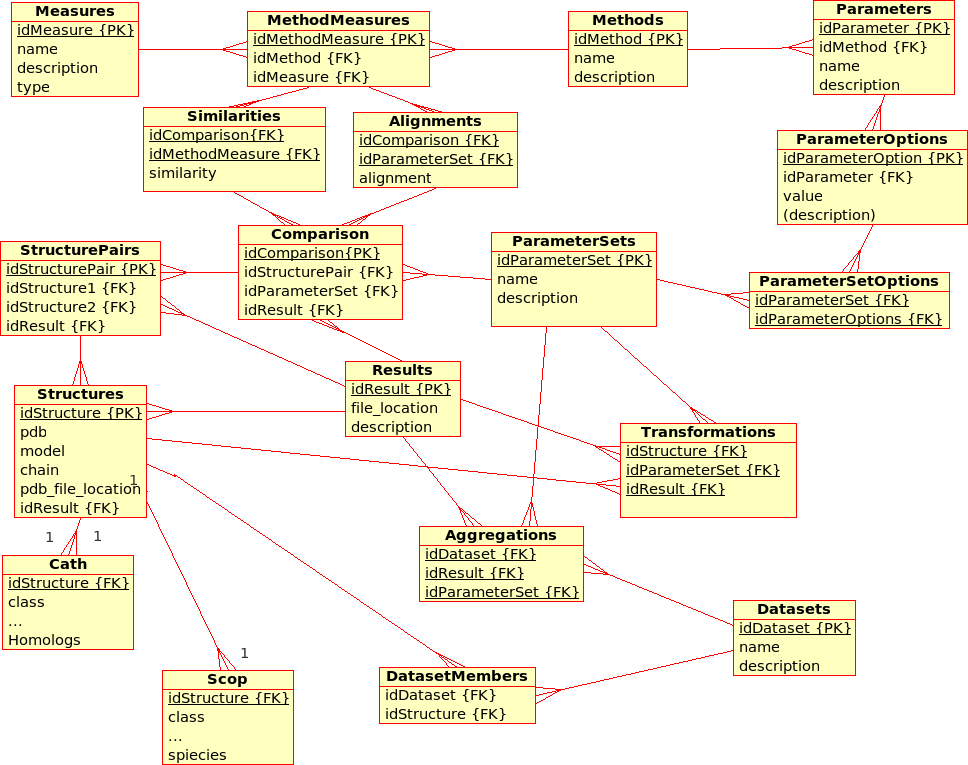
Database Design for the (static) Protein Multiverse¶
The database stores results from Transformations, Comparisons and Compositions:- A Transformation is a process that derives ONE (main) Result from ONE single input file.
Example: The transformation of Structure, Tree, SimilarityMatrix, etc., using a certain Method with a certain ParameterSet, produces a contact map, a tree, ... - A Comparison is a process that derives ONE (main) Result from TWO input files.
Example: The comparison of Structures, Trees, etc., using a Method with a certain ParameterSet, produces a similarity value and an alignment - An Aggregation is a process that derive ONE (main) Result from SEVERAL input files that are grouped together into DataSets.
Example: The aggregation of SimilarityMatrices, Trees, using a Method with a certain ParameterSet, produces a consensus similarity matrix, a consensus tree, ...
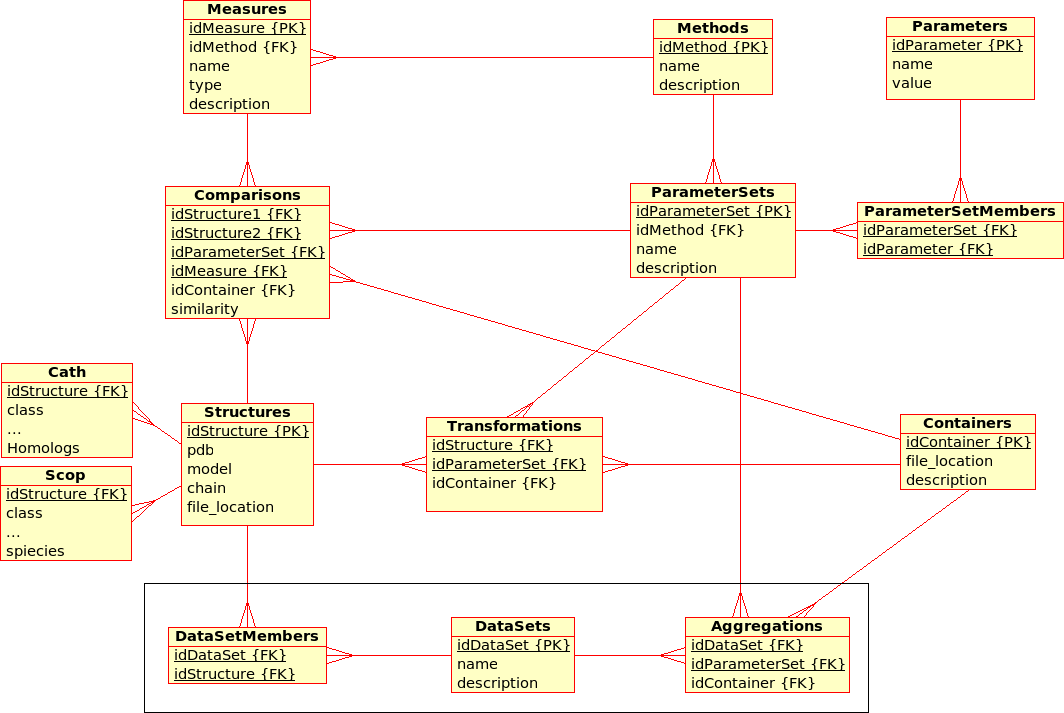
- There are multiple (similarity comparison) Methods: e.g. USM, MaxCMO, DaliLite, ...
- Each Method is executed with a specific ParameterSet, which is a combination of different Parameters with its values: e.g. MaxCMO/restarts/10, USM/compressor/bzip2, ...
- If a Method does not accept any Parameters, the ParameterSet does exist but is empty; e.g. DaliLite, CE, ...
- Each Method procudes multiple similarity Measures: e.g. DaliLite/Z, FAST/Z, MaxCMO/Overlap, ...
- Each Structure is uniquely determined by its PDB code, model and chain. (Domains are not taken into accout yet.) The location of the PDB file is given and a link to a further Container file that holds further information in XML format: e.g. sequence, secondary structure, experimental resolution, ...
- Each Structure is extended by further classifiction information from CATH and SCOP in separtate relations.
- Multiple Structures can be grouped together into DataSets, which are needed for Aggregations.
- The location of the Containers in which results are stored can be found in the Transformations, Comparisons, and Aggregations relations, respectively.
- Additionally, similarity values from Comparisons are stored directly in the database for quicker access. Alignments could be accessed in the same way, as soon as a standardised format has been defined.
Note that this design does not allow Datasets to comprise other files than Structures although some of the Results need to be grouped into a DataSet, too.
Example: Contact maps that have been produces by a Transformation of Structures and that are available from within the Containers need to form a DataSet in order to act as input for the Comparisons with the USM or MaxCMO Methods.
Storing Further Information and Results externally¶
Similarity values are stored directly in the relational database. All further information regarding one structure (e.g. sequence, resolution, ...) or regarding a pair of structures (e.g. alignment, rotation/translation matrices, ...) are stored in external files.For storing further information for single structures, there are several approaches:
- All information in one file: file too big
- All information in separate files grouped by the protein structure
- All information in separate files grouped by methods: files too big
- All information in separate files grouped by pairs: too many files
- All information in separate files grouped by the the first structure: files with unbalanced sizes
- All information in separate files with fixed size:
"Bin-packing" algorithm decides where to put new information, and opens a new "bin" if necessary. "Bins" must be balanced from time to time in order to provide a fast retrieval of information.
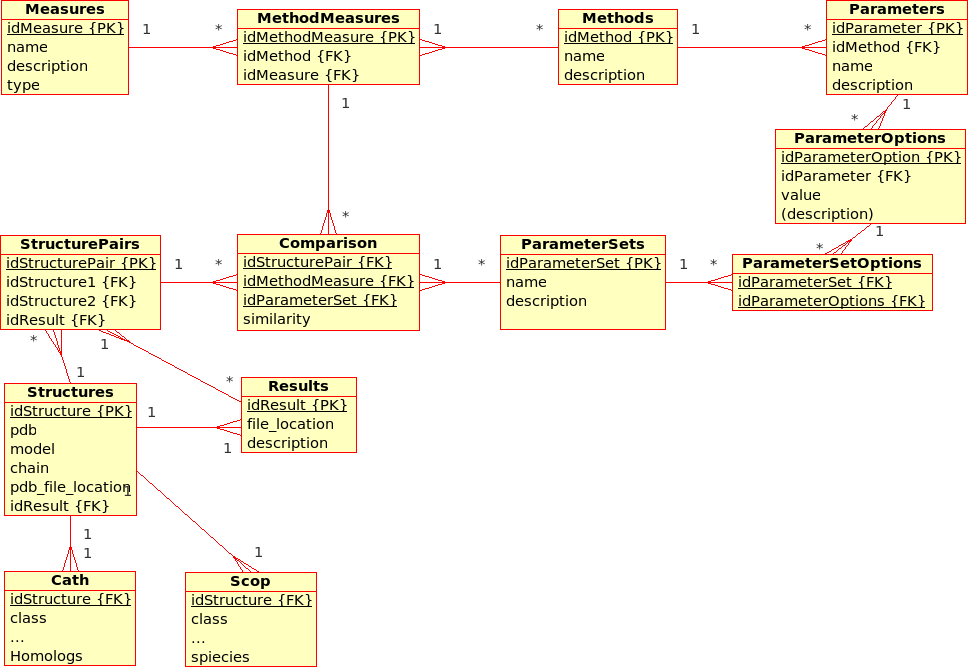
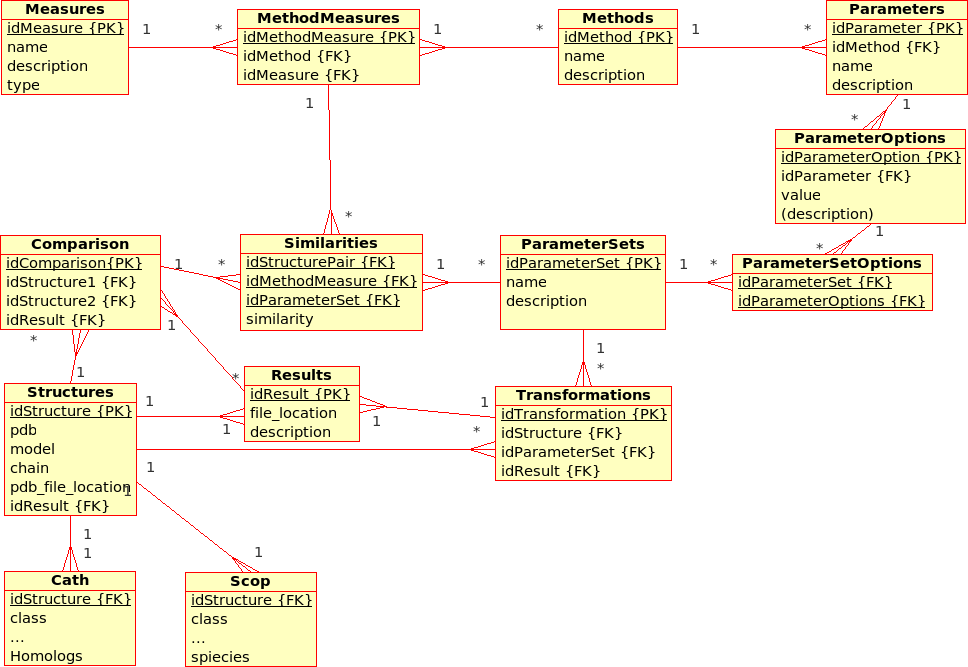
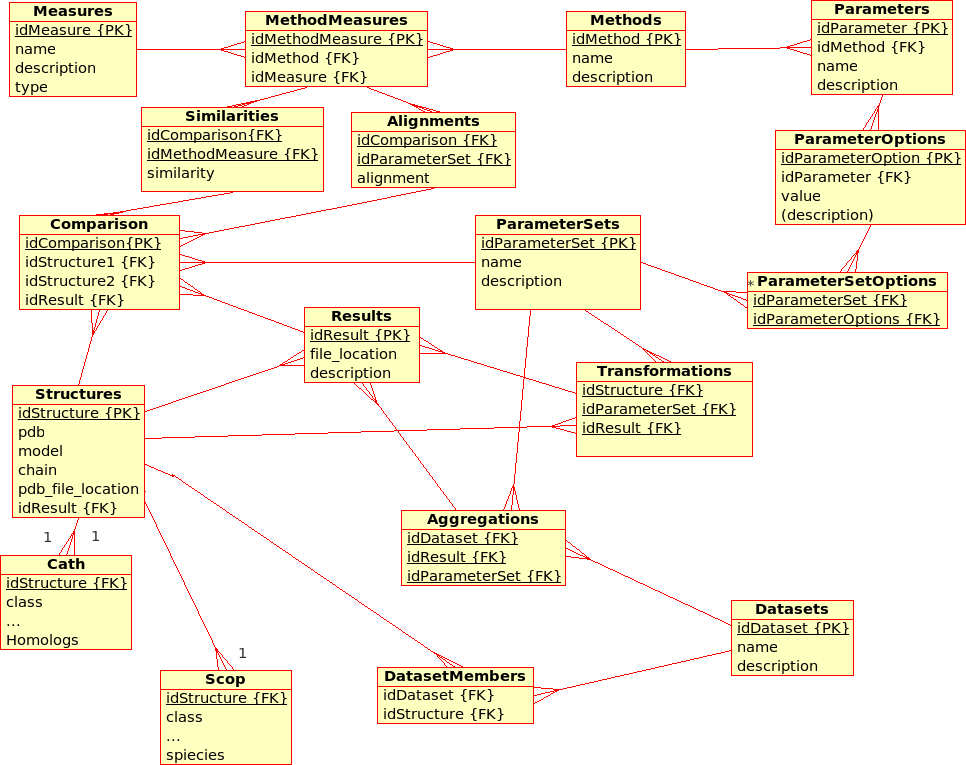
Extended Database Design for (dynamic) Management of Experiments (ProCKSI)¶
This has not been modelled yet, but the database for the (static) Protein Multiverse was designed with the ProCKSI integration in mind.
Some remarks:- Experiments (formerly Requests) apply Methods to_DataSet_ with a certain ParameterSet.
- Packages (formerly Jobs) deal with a subset of a DataSet and a subset of the requested Methods, partitioning the the 3D problem space, and are calculated using the ProCKSI's "stand-alone" core application "in one go". If they are sent to a queuing system, they become a Job there.
- It has to be discussed if there is still the need of a Tasks relation in the database, which have always been rather !RequestMethods.
{kind=link}
{kind=link}
{kind=link}
{kind=link}