DataStorage » History » Version 13
« Previous -
Version 13/16
(diff) -
Next » -
Current version
Anonymous, 10/28/2007 01:04 AM
= Data Storage =
This page describes the design of the database that is/will be used in order to store all necessary pieces information that are obtained from the "stand-alone" ProCKSI ''core'' application (see [wiki:DataStandardisation]).
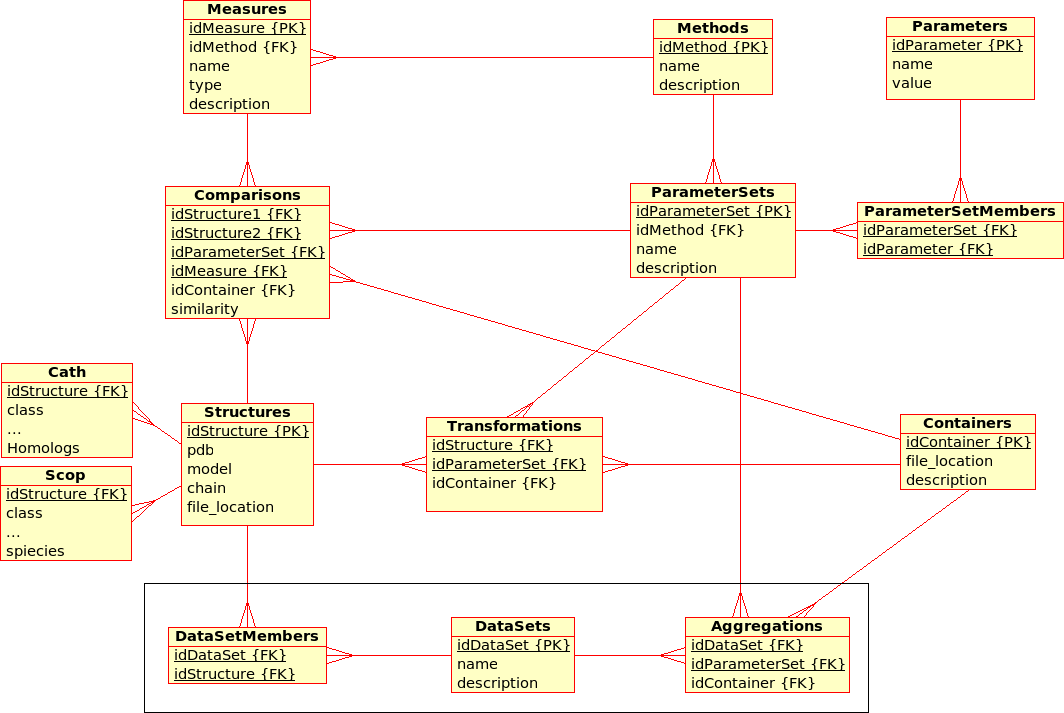
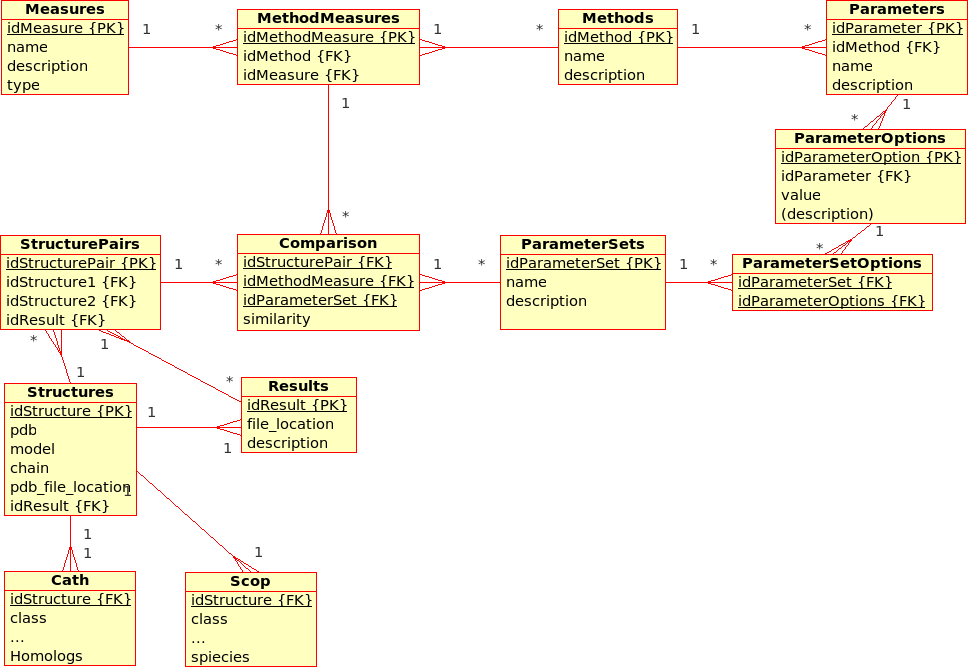
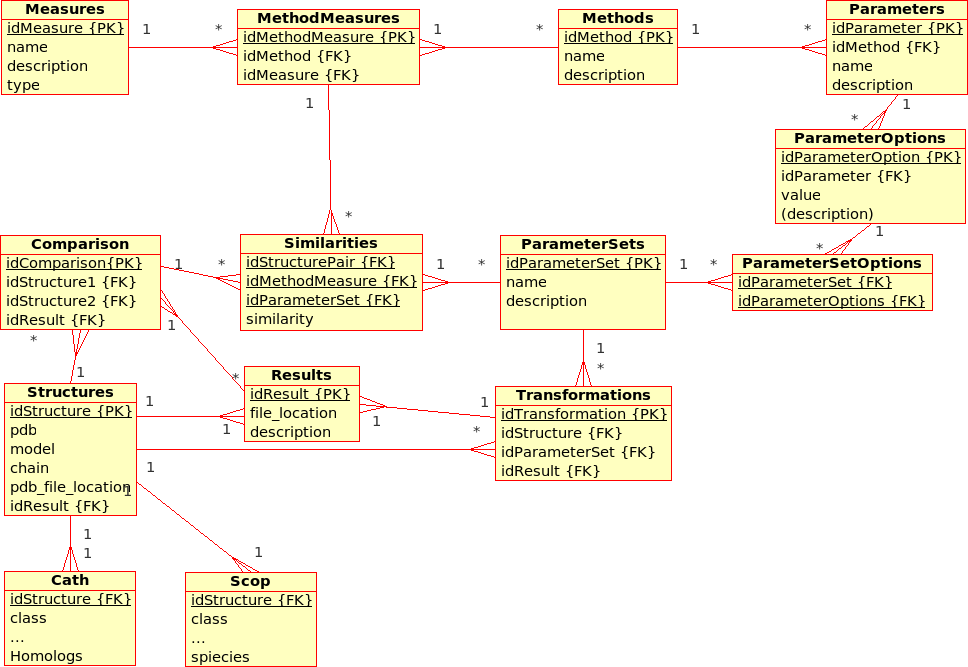
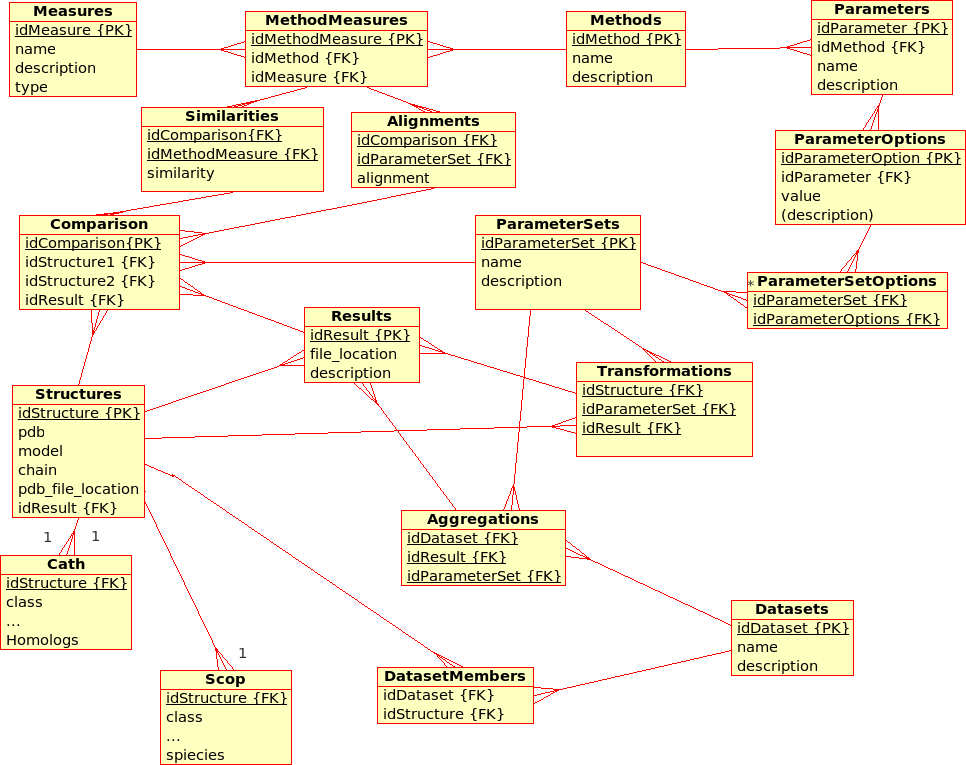
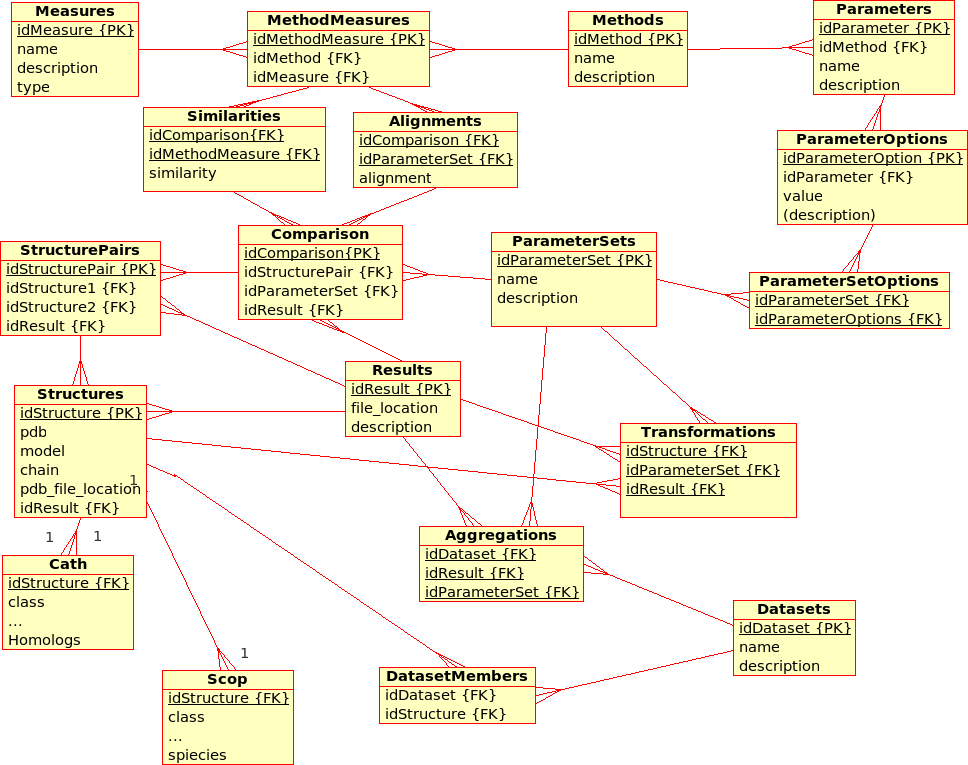
Database Design for the (static) Protein MultiverseThe database stores results from ''Transformations'', ''Comparisons'' and ''Compositions'': * A ''Transformation'' is a process that derives ONE (main) ''Result'' from ONE single input file.br
Example: The transformation of ''Structure'', ''Tree'', ''!SimilarityMatrix'', etc., using a certain ''Method'' with a certain ''!ParameterSet'', produces a contact map, a tree, ... * A ''Comparison'' is a process that derives ONE (main) ''Result'' from TWO input files. br
Example The comparison of ''Structures'', ''Trees'', etc., using a ''Method'' with a certain ''!ParameterSet'', produces a similarity value and an alignment * An ''Aggregation'' is a process that derive ONE (main) ''Result'' from SEVERAL input files that are grouped together into ''!DataSets''. br
Example The aggregation of ''!SimilarityMatrices'', ''Trees'', using a ''Method'' with a certain ''!ParameterSet'', produces a consensus similarity matrix, a consensus tree, ...
Image(ProteinMultiverseDataBase6.png)
- There are multiple (similarity comparison) ''Methods'': e.g. USM, MaxCMO, !DaliLite, ...
- Each ''Method'' is executed with a specific ''!ParameterSet'', which is a combination of different ''Parameters'' with its values: e.g. MaxCMO/restarts/10, USM/compressor/bzip2, ...
- If a ''Method'' does not accept any ''Parameters'', the ''!ParameterSet'' does exist but is empty; e.g. !DaliLite, CE, ...
- Each ''Method'' procudes multiple similarity ''Measures'': e.g. !DaliLite/Z, FAST/Z, MaxCMO/Overlap, ...
- Each ''Structure'' is uniquely determined by its PDB code, model and chain. (Domains are not taken into accout yet.) The location of the PDB file is given and a link to a further ''Container'' file that holds further information in XML format: e.g. sequence, secondary structure, experimental resolution, ...
- Each ''Structure'' is extended by further classifiction information from ''CATH'' and ''SCOP'' in separtate relations.
- Multiple ''Structures'' can be grouped together into ''!DataSets'', which are needed for ''Aggregations''.
- The location of the ''Containers'' in which results are stored can be found in the ''Transformations'', ''Comparisons'', and ''Aggregations'' relations, respectively.
- Additionally, similarity values from ''Comparisons'' are stored directly in the database for quicker access. Alignments could be accessed in the same way, as soon as a standardised format has been defined.
Note that this design does not allow ''Datasets'' to comprise other files than ''Structures'' although some of the ''Results'' need to be grouped into a ''!DataSet'', too.br
Example Contact maps that have been produces by a ''Transformation'' of ''Structures'' and that are available from within the ''Containers'' need to form a ''!DataSet'' in order to act as input for the ''Comparisons'' with the USM or MaxCMO ''Methods''.
=== Storing Further Information and Results externally ===
Similarity values are stored directly in the relational database. All further information regarding one structure (e.g. sequence, resolution, ...) or regarding a pair of structures (e.g. alignment, rotation/translation matrices, ...) are stored in external files.br
For storing further information for ''single structures'', there are several approaches:
* All information in one file: file too big
* All information in separate files grouped by the protein structure
For storing further information for ''pairs of structures'', there are several approaches:
* All information in separate files grouped by methods: files too big
* All information in separate files grouped by pairs: too many files
* All information in separate files grouped by the the first structure: files with unbalanced sizes
* All information in separate files with fixed size:br
"Bin-packing" algorithm decides where to put new information, and opens a new "bin" if necessary. "Bins" must be balanced from time to time in order to provide a fast retrieval of information.
This has not been modelled yet, but the database for the (static) Protein Multiverse was designed with the ProCKSI integration in mind.
Some remarks: * ''Experiments'' (formerly ''Requests'') apply ''Methods'' to''!DataSet'' with a certain ''!ParameterSet''. * ''Packages'' (formerly ''Jobs'') deal with a subset of a ''!DataSet'' and a subset of the requested ''Methods'', partitioning the the 3D problem space, and are calculated using the ProCKSI's "stand-alone" core application "in one go". If they are sent to a queuing system, they become a ''Job'' there. * It has to be discussed if there is still the need of a ''Tasks'' relation in the database, which have always been rather ''!RequestMethods''.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}